Molecular Methods 101
Table of Content
- Pyrosequencing
- RFLP
- Chain Terminators (Sanger Method)
- ASO
- Manual Sequence Analysis
- Automated Sequence Analysis
A. Separation and Detection
1. NUCLEIC ACID PURIFICATION
Extraction and isolation of nucleic acids (DNA and/or RNA) is the key first step before subsequent molecular analysis. DNA subtypes include genomic, plasmid, viral and DNA fragments. RNA subtypes include mRNA, total RNA, viral RNA and RNA fragments.
Protocols vary and are dependent on the type of specimen, required yield, purity and size of the nucleic acids, ease of operation and throughput. Most protocols are categorized as either liquid- or solid-phase extraction but share some common steps such as cell lysis, achieved through the use of detergents, enzymes or physical disruption, and alcohol precipitation which purifies and concentrates the DNA.
1a) Liquid phase extraction is favored when large quantities of DNA or large sample volumes are involved. Removal of cellular proteins and separation of genomic DNA through organic solvent extraction (phenol-chloroform protocol is most well known) allows the isolation of the DNA with the removal of excess cellular proteins. The disadvantages of this method include the number of manual steps, the unintentional loss of the DNA pellet and the hazardous chemicals used.
1b) Solid phase extraction is generally based on the theory that the negatively charged DNA will bind reversibly to positively charged substances such as silica (coated onto membrane filters, columns or magnetic particles) in the presence of chaotropic salts, such as guanidine thiocynate and alcohol. Excess cellular material and proteins are washed away and the DNA is then eluted from the binding matrix. This method is more commonly used because of its relative ease of operation, batch processing, high reproducibility and adaptability to automation.
Agencourt® GenFind® v2 Extraction Process Overview
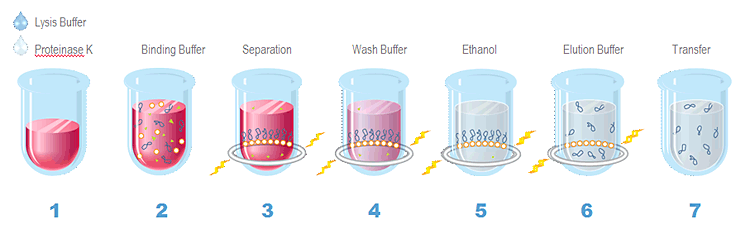
1. Lyse whole blood or serum in Lysis Buffer and Proteinase K 2. Bind genomic DNA to paramagnetic beads 3. Separate beads from contaminants 4. Wash the magnetic beads with Wash Buffer 1 to remove contaminants 5. Wash the magnetic beads with Wash Buffer 2 to remove contaminants 6. Elute DNA from magnetic particles 7. Transfer to new plate
References:
- Image -http://www.agencourt.com/products/spri_reagents/genfindv2/
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- https://www.roche-applied-science.com/PROD_INF/MANUALS/napi_man/napi.htm
- Text- http://acad.erskine.edu/facultyweb/baker/J05-10_DNA%20Technology/Isolation%20of%20Nucleic%20Acids.doc
- Text- GenFind DNA Extraction Kit [package insert]. Madison, WI: Hologic, Inc.; 2009.
2. GEL ELECTROPHORESIS
Gel electrophoresis is a commonly used technique for the separation of deoxyribonucleic acid (DNA), ribonucleic acid (RNA), or protein molecules using an electric current applied to a gel matrix. Separation is based primarily on molecular weight although the physical size and shape of the molecules can also affect results. DNA gel electrophoresis is generally only used after amplification of DNA via PCR. It is usually performed for analytical purposes, but may be used as a preparative technique prior to use of other methods such as mass spectrometry, RFLP, PCR, cloning, DNA sequencing, or Southern blotting for further characterization.
References:
- Animation- http://www.sumanasinc.com/webcontent/ animations/content/gelelectrophoresis.html
- Animation- http://learn.genetics.utah.edu/content/labs/gel/
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- Berg JM, Tymoczko JL Stryer L (2002). Biochemistry(5th ed.). WH Freeman.
3. FLUORESCENCE IN-SITU HYBRIDIZATION
Fluorescence in situ hybridization (FISH) is a cytogenetic technique that can be used for detecting RNA and DNA sequences in cells or tissues. An interphase or metaphase chromosome preparation is produced and placed onto a slide to be denatured. Florescent probes are selected that are complementary to the known sequence. There are four types of probes that are typically used for in situ hybridization: oligonucleotide probes, single stranded DNA probes, double stranded DNA probes and RNA probes (cRNA probes or riboprobes). The probes bind, or hybridize, to areas on the chromosome with a high degree of sequence similarity. The results are then visualized and quantified using a microscope that is capable of exciting the dye and recording images.
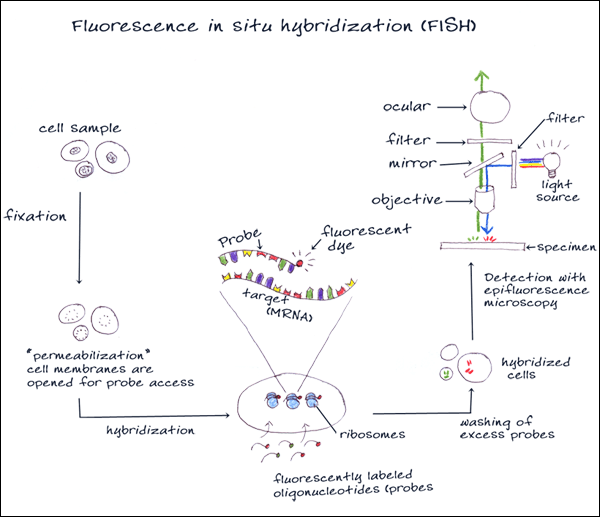
References:
- Image- http://www.bio.davidson.edu/Courses/Molbio/ MolStudents/spring2003/Baxter/MolecularTool.html
- Text- http://science.jrank.org/pages/2775/Fluorescence-in-Situ-Hybridization-FISH.html
- Text- http://www.pathology.washington.edu/galleries/ Cytogallery/main.php?file=fish
4. BLOTTING
Southern blot is a method for probing for the presence of a specific DNA sequence within a non-amplified DNA sample. DNA samples before or after restriction enzyme digestion are separated by gel electrophoresis and then transferred to a membrane by blotting via capillary action (transfer can be increased with vacuum or pressure systems). The membrane is then exposed to a labeled DNA probe that has a complement base sequence to the sequence on the DNA of interest. The resulting fragments can be visualized on film through chemiluminescence or directly on the membrane photometrically. Southern blotting is used commonly to reveal polymorphisms in the DNA sequence as well as large structural alterations, such as deletions, duplications, insertions and rearrangements.
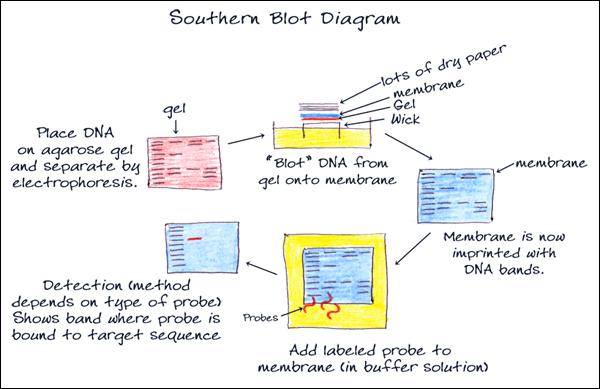
References:
- Animation- http://highered.mcgraw-hill.com/olcweb/cgi/pluginpop.cgi?it=swf::535::535::/sites/dl/free/0072437316/120078/bio_g.swf::Southern%20Blot
- Image- http://askabiologist.asu.edu/expstuff/mamajis/ southern/southern.html
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
B. Nucleic Acid Amplification
1. POLYMERASE CHAIN REACTION
Polymerase chain reaction (PCR) is a technique widely used in molecular biology. PCR amplifies a single or a few copies of a piece of DNA using it as a template for replication, generating millions of copies of a particular DNA sequence. The method relies on thermal cycling with cycles of repeated heating and cooling of the reaction for DNA melting and enzymatic replication. Primers (short DNA fragments) containing sequences complementary to the target region, along with a DNA polymerase (after which the method is named), are key components to enable selective and repeated amplification.
References:
- Animation- http://www.sumanasinc.com/webcontent/ animations/content/pcr.html
- Animation- http://learn.genetics.utah.edu/content/labs/pcr/
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- Joseph Sambrook and David W. Russel (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
1A. PCR VARIATIONS
Real-time polymerase chain reaction (qPCR) uses the general principles of polymerase chain reaction but instead of waiting until the end-point of the reaction to quantify results, the reaction is quantified in “real-time” during amplification cycles. Fluorescent dyes or probes that can signal the relative quantity of DNA are added to the PCR mixture before amplification. Amplification and fluorescence monitoring occur in the same reaction tube with no need for sample transfer, reagent additions or gel separation steps, reducing the risk of specimen contamination.
Reverse transcription polymerase chain reaction (RT-PCR) uses the general principles of polymerase chain reaction to amplify DNA. With RT-PCR, strands of RNA are reverse transcribed to its DNA compliment using the enzyme reverse transcriptase. The complementary DNA can then be amplified by using either traditional or real-time PCR. The exponential amplification via RT-PCR provides for a highly sensitive technique, where a very low copy number of RNA molecules can be detected and is widely used in the diagnosis of genetic diseases and, semi-quantitatively, in the determination of different RNA molecules within a cell or tissue as a measure of gene expression.
References:
- Animation- http://www.bio.davidson.edu/Courses/ immunology/Flash/RT_PCR.html
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- Joseph Sambrook and David W. Russel (2001). Molecular Cloning: A Laboratory Manual (3rd ed.). Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
- Text- Innis MA et al. (1990). “Academic Press”. PCR Protocols: A Guide to Methods and Applications.
2. INVADER® METHOD
A signal amplification method for detection of specific nucleic acid sequences. This method uses two types of isothermal reactions: a primary reaction that occurs on the targeted DNA sequence and a secondary reaction that produces a fluorescent signal. In the primary reaction, two types of sequence specific oligonucleotides (i.e. a probe oligonucleotide and an Invader® oligonucleotide) bind to the DNA target sequence. When these oligonucleotides overlap by at least one base pair on the target sequence, an invasive structure forms that acts as a substrate for the Cleavase® enzyme. The enzyme cleaves the 5′ portion (flap) of the probe at the position of the overlap.
The probes are present in excess and cycle rapidly on and off the target sequence so that many cleaved 5′ flaps are generated per target sequence. The cleaved flaps then bind to a universal hairpin fluorescence resonance energy transfer (FRET) oligonucleotide creating another invasive structure that the Cleavase® enzyme recognizes as a substrate. The enzyme cleaves the FRET oligonucleotides between the fluorophore and quencher molecule and produces fluorescence signal as the cleaved flaps cycle on and off. For each copy of target, the combined primary and secondary reactions result in 106 – 107 fold signal amplification per hour. The flap sequences and FRET oligonucleotides are universal since they are not complementary to the targeted sequence.
- Text- Day S, Mast A. Invader assay. In: Fuchs J. & Podda M., eds. Encyclopedia of Diagnostic Genomics and Proteomics Marcel Dekker, Inc; 2004.
3. HYBRID CAPTURE
Used to detect and classify HPV DNA, this method uses signal amplification technology and probe hybridization. Two separate pools of RNA probes specific for 5 low and 13 high-risk HPV DNA types are used, and results are reported as low-risk or high-risk HPV DNA detected with no indication of the specific type.
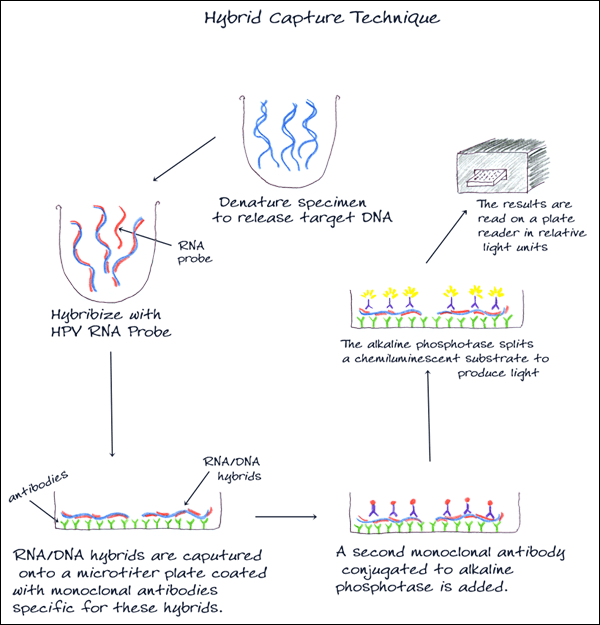
References:
- Image- http://www.papillomavirus.cz/images/hybridcapture.jpg
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- http://www.hpvinformation.com/hcp/Resource-Center/~/media/Files/QIAGENCMS/Corporate/Web/TheHPVTest/Resources/1051669_FLY_HPV_205x275_0708_lr_4.ashx
4. TRANSCRIPTION-BASED AMPLIFICATION METHODS
Transcription-based amplification methods include nucleic acid sequence-based amplification (NASBA), transcription-mediated amplification (TMA), and self-sustained sequence replication (3SR). The process is isothermal and uses the combined actions of reverse transcriptase, RNase H and RNA polymerase. The method may be applied to single-stranded RNA or double-stranded DNA targets. As in PCR, all reagents can be included in one mixture, amplification is exponential and the process can be completed in less than an hour.
In the first step of amplification, the primer hybridizes to the target rRNA at a defined site and reverse transcriptase creates a DNA copy of the target rRNA. The RNA in the resulting duplex is degraded by RNase activity. A second primer then binds to the DNA copy and a new strand of DNA is synthesized, creating a double-stranded DNA molecule. Transcription is initiated and newly synthesized RNA amplicons reenters the TMA process, serving as templates for a new round of replication.
References:
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- http://www.pcr-encyclopedia.com/transcription-mediated-amplification-2252.html
- Text- http://www.chlamydiae.com/restricted/docs/ labtests/diag_2ndtma.asp
5. STRAND DISPLACEMENT AMPLIFICATION
Strand displacement amplification (SDA) is an isothermal nucleic acid amplification method. DNA is heat denatured in the presence of four primers and a modified deoxy-nucleotide and is then combined with two enzymes – an exonuclease-deficient polymerase and a restriction enzyme. The two flanking primers in the amplification process contain a restriction site that gets nicked by the restriction enzyme causing the displacement of strands that then get primed, extended and nicked. Newly synthesized DNA are nicked by a restriction enzyme, polymerase starts amplification again, displacing the newly synthesized strands. 109 copies of DNA can be made in one reaction.
References:
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text-http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=312258&blobtype=pdf
6. LIGASE CHAIN REACTION (LCR)
Ligase chain reaction (LCR) is a method of DNA amplification similar to PCR. LCR differs from PCR because it amplifies the probe molecule rather than producing amplicon through polymerization of nucleotides. Two probes are used per each DNA strand and are ligated together to form a single probe. LCR uses both a DNA polymerase enzyme and a DNA ligase enzyme to drive the reaction. Like PCR, LCR requires a thermal cycler to drive the reaction and each cycle results in a doubling of the target nucleic acid molecule. LCR can have greater specificity than PCR but requires that the exact sequence in the region being amplified is known.
References:
- Text- http://www.biochem.northwestern.edu/holmgren/ Glossary/Definitions/Def-L/ligase_chain_reaction.html
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- Wiedmann M, Wilson WJ, Czajka J, Luo J, Barany F, Batt CA. “Ligase chain reaction (LCR)–overview and applications.” PCR Methods and Applications 1994 Feb;3(4):S51-64 PMID: 8173509
C. DNA Sequence Analysis
1. PYROSEQUENCING
Pyrosequencing is a method that detects nucleic acid sequences of short segments without electrophoresis. Solutions of individual deoxynucleotide triphosphates (dNTP) are added one by one to a PCR-generated single-stranded template in the presence of a primer and enzymes. The pyrophosphate released with a complementary match initiates an enzymatic reaction that results in the release of luminescence. The light produced can be detected by a camera and analyzed.
References:
- Animation- http://www.dnatube.com/video/2954/Pyro-Sequencing
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
2. RESTRICTION FRAGMENT LENGTH POLYMORPHISM (RFLP)
DNA extracted from cells can be cut into shorter fragments using restriction endonucleases. Differences (polymorphisms) between two or more samples of DNA can be detected when the resulting restriction fragments are separated according to their lengths by gel electrophoresis. For example, if a DNA fragment contains a sequence that is recognized, that sequence is cleaved by the restriction enzyme creating 2 fragments. If a mutation of that sequence exists instead, the sequence will not be recognized and cleavage will not occur leaving the original length of DNA. PCR can aid in the detection by amplifying fragments before restriction. Southern blotting can be added as a technique to detect DNA alterations that span large regions. This method has been called Cleaved Amplified Polymorphic Sequence (CAPS).
RFLP analysis continues, but is now usually performed by polymerase chain reaction (PCR) methods. Amplification can be directed across the altered restriction site, and the products digested with the restriction enzyme. Alternatively, the amplified segment can be analyzed by Allele specific oligonucleotide (ASO) probes, a process that can often be done by a simplified blot technique or Dot Blot.
References:
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- http://www.ncbi.nlm.nih.gov/projects/genome/probe/ doc/TechCAPS.shtml
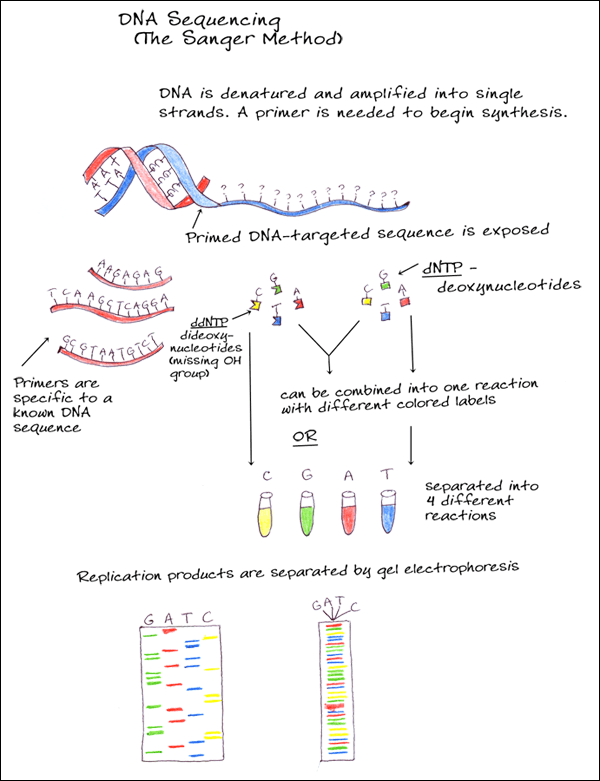
3. CHAIN TERMINATORS (SANGER METHOD)
A single stranded DNA molecule is amplified and primed and then combined with new deoxynucleotides (dNTPs) as well as dideoxy-nucleotides (ddNTPs). The dideoxynucleotides are similar to dNTPs but are missing the hydroxyl group which prevents new bases from being added after it, thus terminating synthesis. Different colored fluorescent tags are added for each of the nucleotides to either the dNTPs or the ddNTPs so that when the DNA is separated according to size during electrophoresis, the sequence can be determined.
Currently, the use of fluorescent rather than radioactive labels and advances in gel electrophoresis, along with automated analysis of results, has made DNA sequencing quicker and more accurate.
References:
- Animation- http://www.dnalc.org/resources/animations/ sangerseq.html
- Text- Bruns, David E, et al. (2007) Fundamentals of Molecular Diagnostics. Saunders
- Text- http://www.scq.ubc.ca/genome-projects-uncovering-the-blueprints-of-biology/
- Text- http://www.bio.davidson.edu/Courses/Molbio/ MolStudents/spring2003/Obenrader/sanger_method_page.htm
4. ALLELE SPECIFIC OLIGONUCLEOTIDE (ASO)
An allele specific oligonucleotide is a short piece of synthetic DNA specific for only one version, or allele, of the DNA being tested and even a single base change will hinder hybridization. To be detected after it has bound to its target, the ASO must be labeled with a radioactive, enzymatic, or fluorescent tag.
- Text-http://en.wikipedia.org/wiki/Allele_specific_oligonucleotide
5. MANUAL SEQUENCE ANALYSIS
The products of the sequence are loaded in four parallel lanes on a gel. Thus every band that shows up in that lane represents the termination of a sequencing product. The gel separates the sequencing products based on size; smaller fragments travel through the gel faster then longer fragments. The four lanes are read together in a horizontal hierarchy from bottom (smallest) to top (largest). The blue line traces the order that the bands are read and the sequence of the fragment is shown on side. The products of DNA sequencing can be visualized because the primer is tagged with a radioactive label. When exposed to a piece of X-ray film, the radioactivity exposes the film showing up as a dark band. The sequence is then read from the film by the researcher or technician.
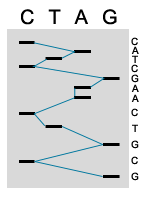
- Text- http://www.uvm.edu/~cgep/Education/Sequence.html
6. AUTOMATED SEQUENCE ANALYSIS
In automated sequencing, the products are labeled with a fluorescent dye rather then a radioactive label. Each fragment has a different color at its end depending on which is the terminating nucleotide (ddNTP). This allows the products of sequencing to be run on a single lane of a gel rather then in four parallel lanes. In addition, the sequencing of nucleotides is determined by the computer, rather then being read manually by a technician. As the samples pass through the gel, a laser excites the fluorescent labels. A computer collects and analyzes this data, reading the sequence of the DNA. Thus automated sequencing is much faster and more efficient then manual sequencing.
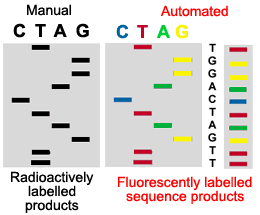
This figure illustrates why automated sequenced products can be run in a single lane of a gel, while manually sequenced products must be run in four adjacent lanes of a gel.
References:
- Text- http://www.uvm.edu/~cgep/Education/Sequence.html
Click here to return to the main menu